jbrowse-plugin-mafviewer
A viewer for multiple alignment format (MAF) files in JBrowse 2
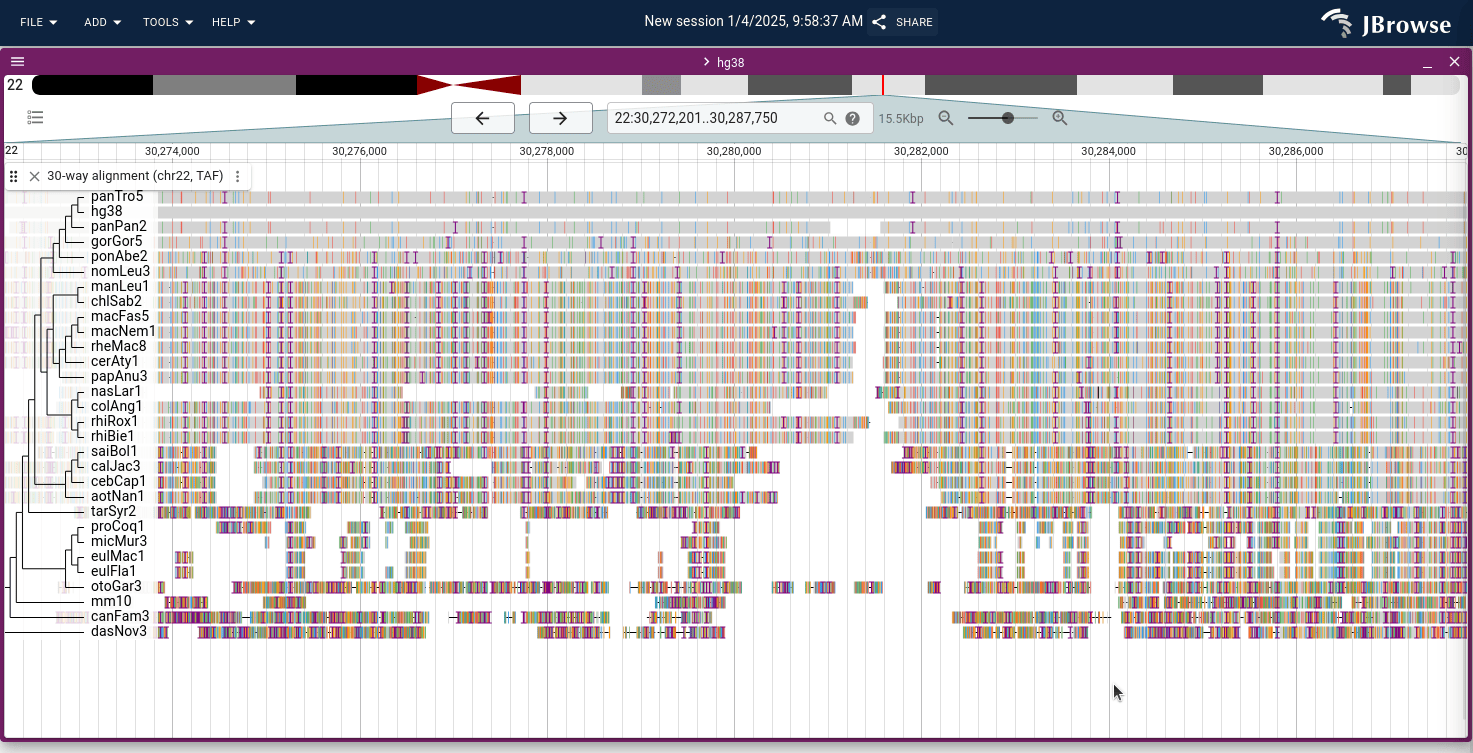
Demo
Supported formats
This plugin supports three input formats:
- BigMaf - UCSC BigMaf format e.g. .bb/.bigMaf (https://genome.ucsc.edu/goldenpath/help/bigMaf.html)
- MAF tabix - bgzip-compressed MAF converted to BED format with tabix index
- TAF (Taffy) - bgzip-compressed TAF format (https://github.com/ComparativeGenomicsToolkit/taffy)
GUI usage (e.g. in JBrowse Desktop)
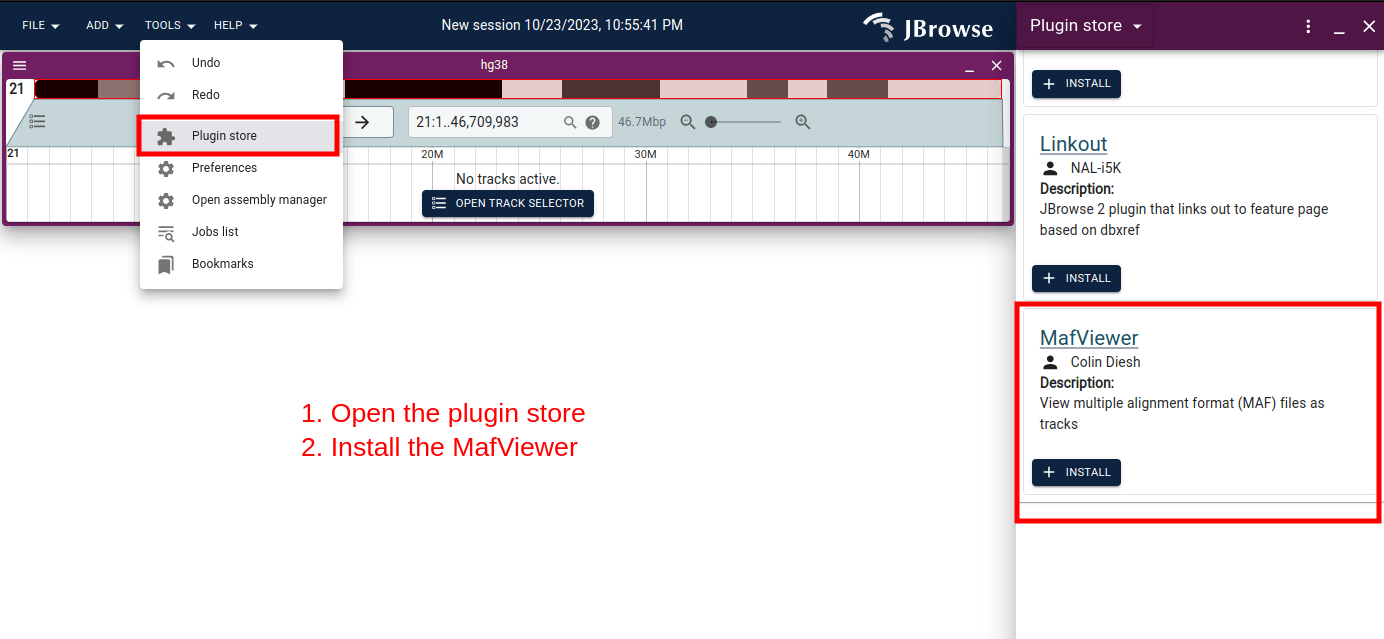
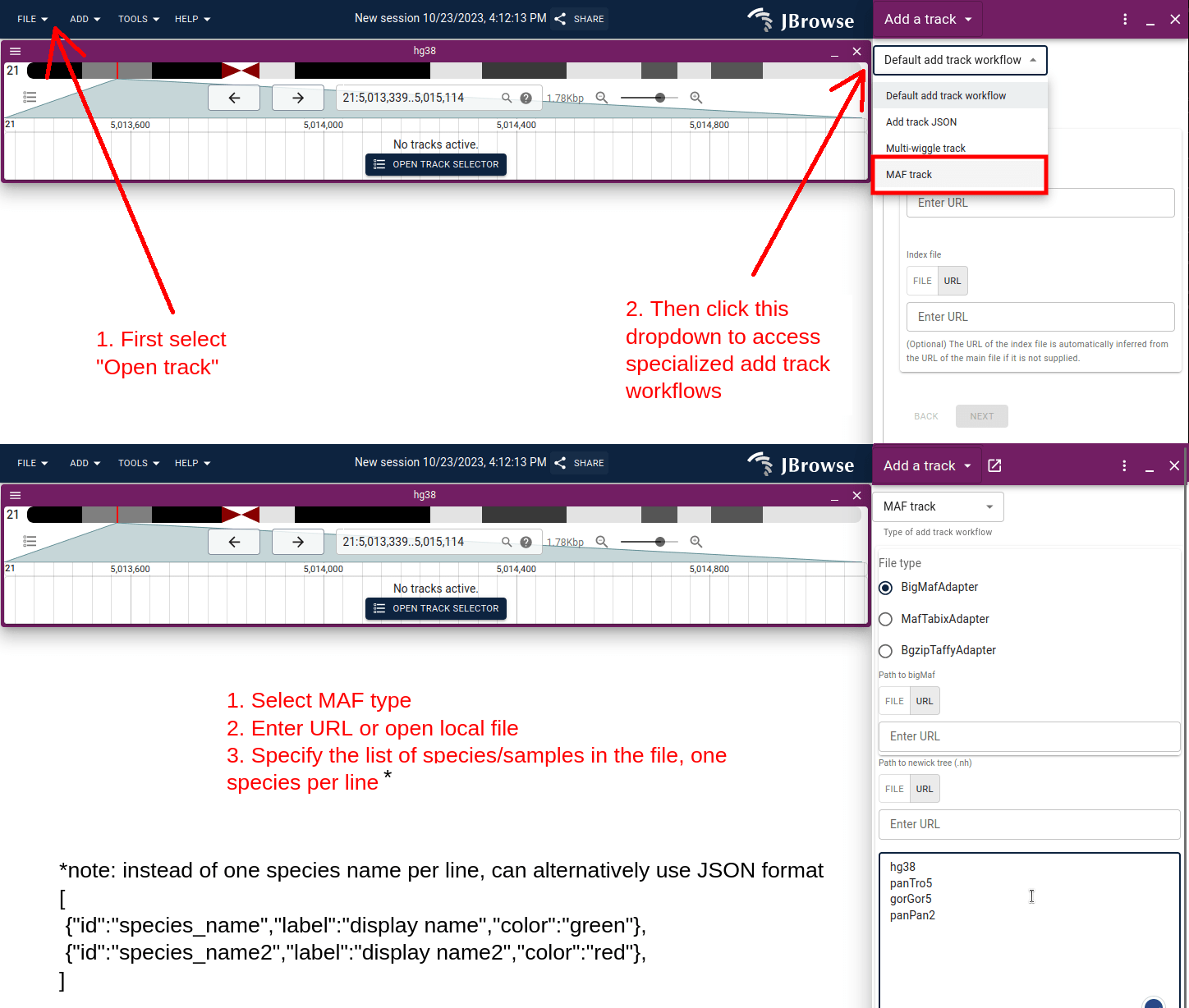
This short screenshot workflow shows how you can load your own custom MAF files via the GUI.
First install the plugin via the plugin store:
Then use the custom "Add track workflow":
Manual config entry
Add plugin to your jbrowse 2 config.json
{
"plugins": [
{
"name": "MafViewer",
"url": "https://jbrowse.org/plugins/jbrowse-plugin-mafviewer/dist/jbrowse-plugin-mafviewer.umd.production.min.js"
}
]
}Example BgzipTaffyAdapter config (TAF format)
TAF (Transposed Alignment Format) is often more compact than MAF and works well with large multi-species alignments. See https://github.com/ComparativeGenomicsToolkit/taffy for more details.
{
"type": "MafTrack",
"trackId": "taf-example",
"name": "TAF alignment",
"assemblyNames": ["hg38"],
"adapter": {
"type": "BgzipTaffyAdapter",
"tafGzLocation": {
"uri": "alignment.taf.gz"
},
"taiLocation": {
"uri": "alignment.taf.gz.tai"
},
"nhLocation": {
"uri": "species_tree.nh"
}
}
}The nhLocation is optional but provides a species tree for display in the
sidebar.
Example MafTabixAdapter config
You can use nhLocation (newick tree) or samples array on the adapter.
{
"type": "MafTrack",
"trackId": "chrI.bed",
"name": "chrI.bed",
"adapter": {
"type": "MafTabixAdapter",
"samples": ["ce10", "cb4", "caeSp111", "caeRem4", "caeJap4", "caePb3"],
"bedGzLocation": {
"uri": "chrI.bed.gz"
},
"index": {
"location": {
"uri": "chrI.bed.gz.tbi"
}
}
},
"assemblyNames": ["c_elegans"]
}Example BigMafAdapter config
You can use nhLocation (newick tree) or samples array on the adapter.
{
"type": "MafTrack",
"trackId": "bigMaf",
"name": "bigMaf (chr22_KI270731v1_random)",
"adapter": {
"type": "BigMafAdapter",
"samples": [
"hg38",
"panTro4",
"rheMac3",
"mm10",
"rn5",
"canFam3",
"monDom5"
],
"bigBedLocation": {
"uri": "bigMaf.bb"
}
},
"assemblyNames": ["hg38"]
}Example with customized sample names and colors
{
"trackId": "MAF",
"name": "example",
"type": "MafTrack",
"assemblyNames": ["hg38"],
"adapter": {
"type": "MafTabixAdapter",
"bedGzLocation": {
"uri": "data.txt.gz"
},
"index": {
"location": {
"uri": "data.txt.gz.tbi"
}
},
"samples": [
{
"id": "hg38",
"label": "Human",
"color": "rgba(255,255,255,0.7)"
},
{
"id": "panTro4",
"label": "Chimp",
"color": "rgba(255,0,0,0.7)"
},
{
"id": "gorGor3",
"label": "Gorilla",
"color": "rgba(0,0,255,0.7)"
},
{
"id": "ponAbe2",
"label": "Orangutan",
"color": "rgba(255,255,255,0.7)"
}
]
}
}The samples array is either string[]|{id:string,label:string,color?:string}[]
Creating MAF files as Cactus pangenome/HAL
You can create a MAF file from a Cactus pangenome graph using ComparativeGenomeToolkit
This page discusses some examples
https://github.com/ComparativeGenomicsToolkit/cactus/blob/master/doc/progressive.md#maf-export
Thanks to Sam Talbot (https://github.com/SamCT) for initially creating the Cactus -> JBrowse 2 MAF example.
Note: This plugin expects non-overlapping alignment blocks. Ensure your MAF/TAF file meets this requirement.
Prepare data
Three formats are supported:
- BigMaf - UCSC BigMaf format (see https://genome.ucsc.edu/FAQ/FAQformat.html#format9.3)
- MAF tabix - bgzip-compressed BED converted from MAF using maf2bed
- TAF (Taffy) - bgzip-compressed TAF with .tai index (recommended for large alignments)
Note: All formats start with a MAF as input. Your MAF file should contain the species name and chromosome name (e.g. hg38.chr1) in the sequence identifiers.
Option 1. Preparing BigMaf
Follow instructions from https://genome.ucsc.edu/FAQ/FAQformat.html#format9.3
Option 2. Preparing MAF tabix
Start by converting the MAF into a pseudo-BED format using the maf2bed tool
# from https://github.com/cmdcolin/maf2bed
cargo install maf2bed
cat file.maf | maf2bed hg38 | sort -k1,1 -k2,2n | bgzip > out.bed.gz
tabix out.bed.gzThe second argument to maf2bed is the genome version e.g. hg38 used for the main species in the MAF (if your MAF comes from a pipeline like Ensembl or UCSC, the identifiers in the MAF file will say something like hg38.chr1, therefore, the argument to maf2bed should just be hg38 to remove hg38 part of the identifier.
If your MAF file does not include the species name as part of the identifier, you should add the species name to the scaffold/chromosome names (e.g. create hg38.chr1 if it was just chr1 before).
If all is well, your BED file should have 6 columns, with
chr, start, end, id, score, alignment_data, where alignment_data is
separated between each species by ; and each field in the alignment is
separated by :.
Note: If you can't use the cargo install maf2bed binary, there is a
bin/maf2bed.pl perl version of it in this repo
Option 3. Preparing TAF (Taffy)
TAF (Transposed Alignment Format) is a column-oriented format that is often more compact than MAF, especially for large multi-species alignments. It uses run-length encoding and avoids the block fragmentation issues that MAF has with many sequences.
Install taffy from https://github.com/ComparativeGenomicsToolkit/taffy
# Convert MAF to bgzip-compressed TAF and create index
taffy view -i file.maf | bgzip > out.taf.gz
taffy index -i out.taf.gzThis creates out.taf.gz and out.taf.gz.tai (the index file).
For large alignments, you may want to normalize the TAF file first to merge short alignment blocks:
taffy view -i file.maf | taffy norm | bgzip > out.taf.gz
taffy index -i out.taf.gzFootnote
This is a port of the JBrowse 1 plugin https://github.com/cmdcolin/mafviewer to JBrowse 2